Imputing Missing Genomes using Computational Methods
Link to Research Lab Link to Project Wiki
![]()
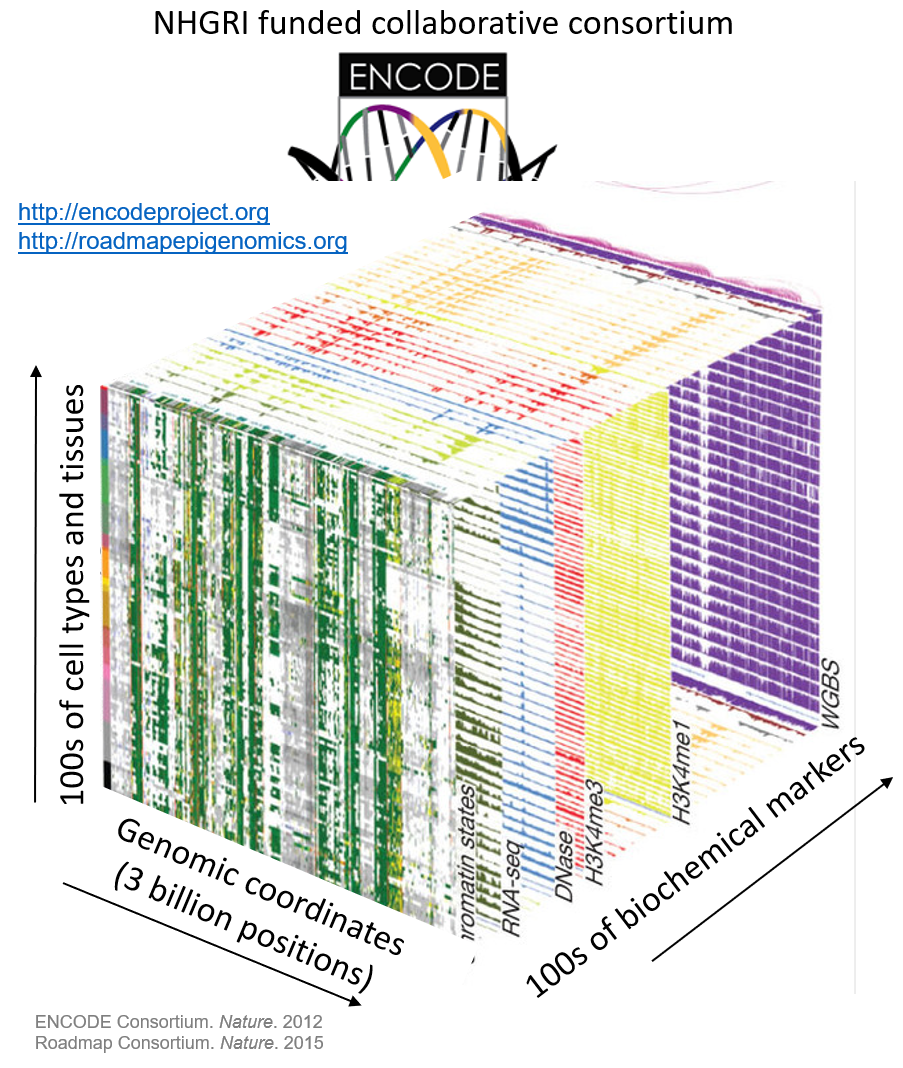
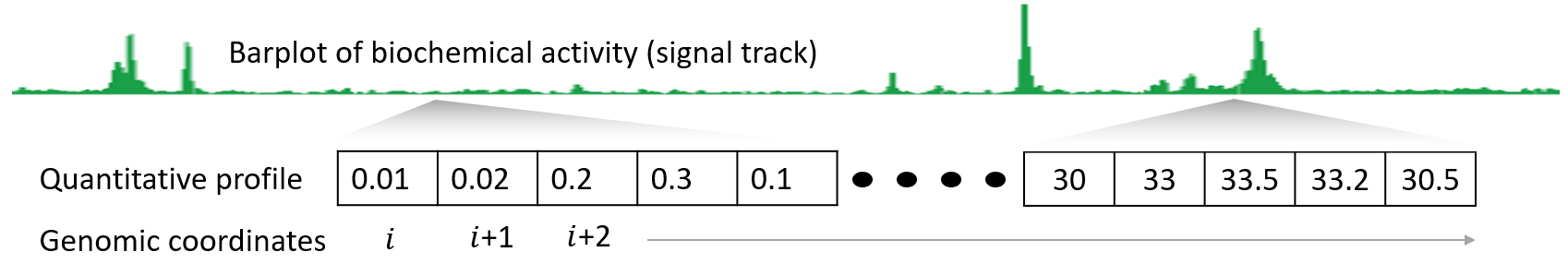
The Encyclopedia of DNA Elements (ENCODE) Consortium is organizing a challenge to accurately impute biochemical data associated with functional genomic elements in a variety of cell types. The consortium has performed many experiments to characterize biochemical activity associated with regulatory activity across the entire human genome in diverse cell types and tissues. However, many combinations of assays and cell types remain to be performed. These experiments are expensive, and technical challenges may prevent comprehensive characterization of all marks in all cell types, so computational methods capable of predicting the output of these assays are potentially valuable. The goal of this research is to develop an accurate and interpretable computational method to impute missing ATAC-seq, DNase-seq, and histone modification ChIP-seq data in various cell types.
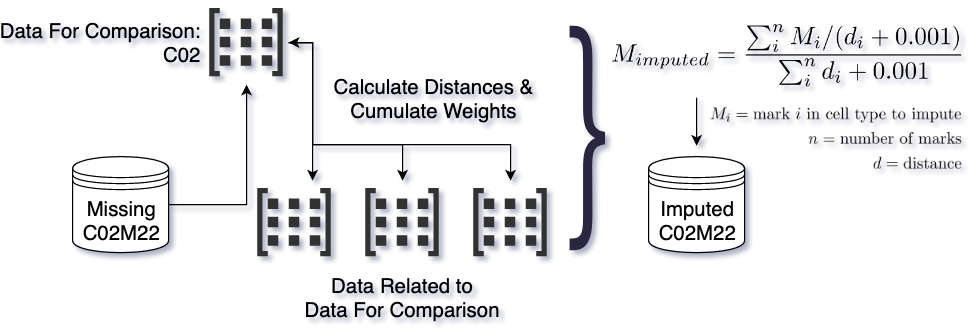
For each combination of cell type and assay to impute, we used both the same assay in other cell types and other assays in the same cell type. First we divided the genome, which was initially binned at 25-bp resolution, into larger non-overlapping windows. For each window we calculated the distance to each other cell type from the cell type to impute, using the assays available for the cell type to impute.
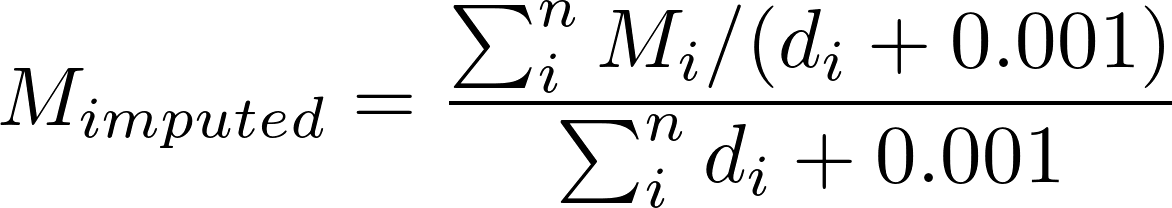
For each bin within the window, we calculated the average of the value of the assay to impute in the other cell types, weighted by the cell type distance for the entire window.
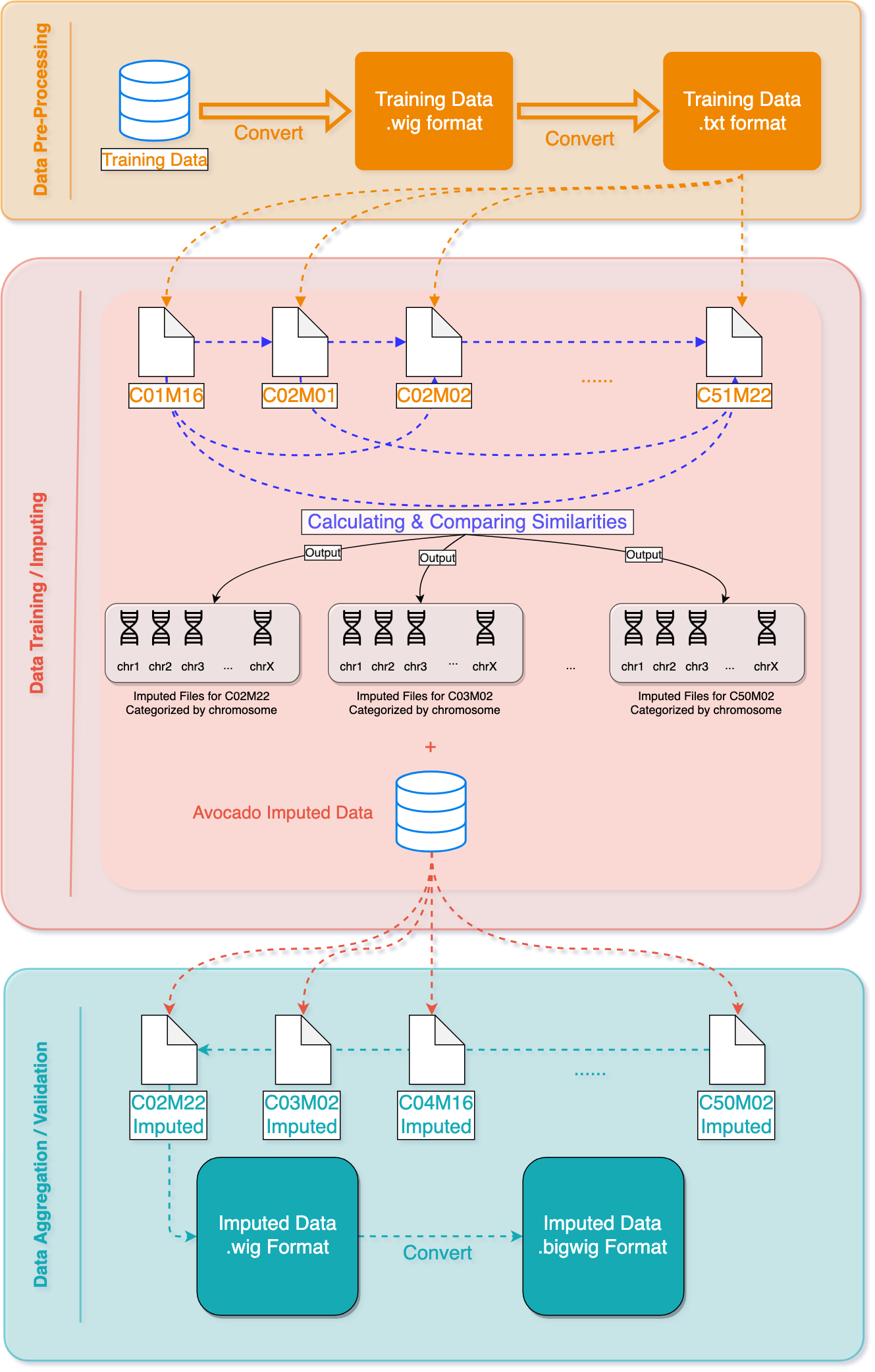
We also used the inverse approach: for each window we calculated the distance to each other assay from the assay to impute, using the cell types available for the assay to impute. We then calculated the average of the other assays in the cell type to impute, weighted by the assay distance. Finally, we calculated the mean of the cell-type-distance imputation and the assay-distance imputation.
We tested a variety of distance metrics and window sizes using the performance metrics provided by the imputation challenge. The cityblock (L1) distance metric and window size of 30 bins (750 bp) performed best. The results from scoring statistics showed that we improved the accuracy 38% comparing to the baseline, which is the average prediction over the entire genome, on average.
Sample Imputation Result
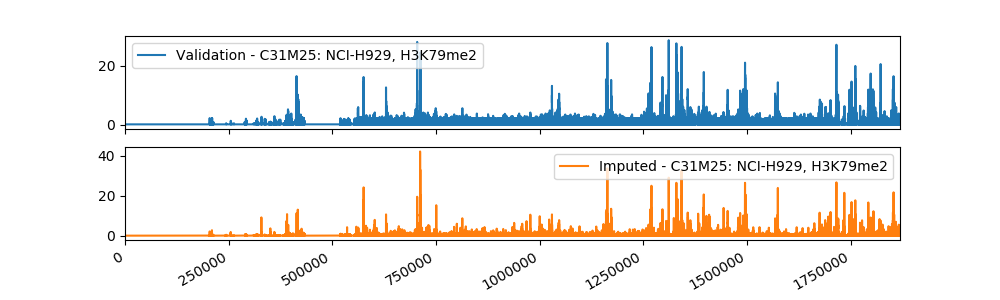
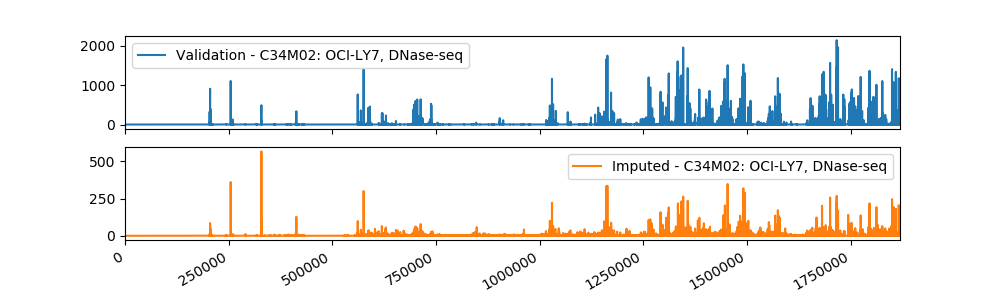
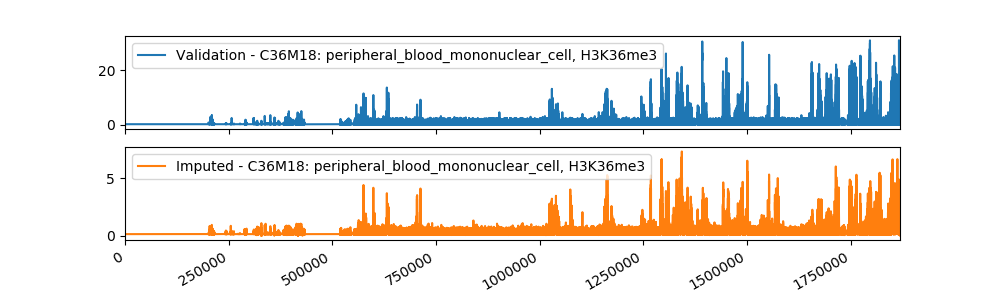
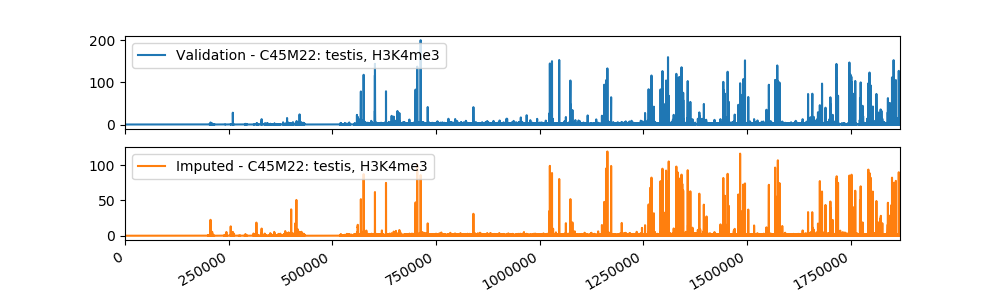
The current imputed files have been tested using different evaluation metrics provided by ENCODE, and the results showed that our method is more accurate than the baseline approach of taking the mean of the assay in other cell types. We plan to use a machine learning algorithm that takes as input our imputations and the imputations from Avocado, a deep neural network tensor factorization method, to produce the final imputation.